





Лимфопролиферативные заболевания, особенно В-клеточные лейкозы – вялотекущие. Чаще при них используется тактика «наблюдай и жди». У некоторых пациентов эта патология может осложниться синдромом Рихтера. По каким-либо причинам параллельно имеющемуся лимфолейкозу у пациентов развивается вторая диффузная крупноклеточная опухоль. Проявляется патология ухудшением общего состояний, появлением симптомов развития крупноклеточной опухоли. В большинстве случаев синдром Рихтера ухудшает прогноз для жизни пациента, даже при использовании адекватной химиотерапии.
Причины развития синдрома Рихтера
Один из предрасполагающих к развитию синдрома Рихтера факторов - инфицирование организма вирусом Эпштейна-Барр.Впервые случай возникновения крупноклеточной диффузной опухоли у пациента с хроническим был описан Рихтером ещё в 1928 году. До сих пор достоверно не известно, является ли возникшая опухоль клоном первичной, или же это два независимо возникших злокачественных образования. По данным иммунологических и генетических исследований, в 1/3 случаев диффузная крупноклеточная опухоль развивается из клональных клеток зрелоклеточной лимфомы, а в 2/3 случаев их взаимосвязь не доказана. Соответственно и точной причины, почему у некоторых пациентов развивается вторая опухоль, достоверно не известно. Считается, что предрасполагающими факторами являются:
- Вирус. При исследовании крупноклеточных лимфом у пациентов с синдромом Рихтера было выявлено ДНК , являющегося предрасполагающим фактором развития и других типов лимфом.
- Иммуносупрессия из-за интенсивной химеотерапии. Частота развития синдрома Рихтера у больных хроническим лимфолейкозом увеличивается при приёме флударабина. При приёме пуриновых аналогов такая закономерность не обнаружена.
- Прогрессирование лимфопролиферативного заболевания. Некоторые исследователи считают, что синдром Рихтера является следствием развития хронического лимфолейкоза. Возникает у пациентов с индивидуальной предрасположенностью к такому осложнению.
Чтобы подтвердить или опровергнуть эти причины развития синдрома Рихтера, проведённых исследований недостаточно.
К тому же встречается синдром Рихтера крайне редко. Он развивается у 3-10 % пациентов со зрелоклеточными лимфопролиферативными патологиями. Появление крупноклеточной лимфомы ухудшает состояние пациента и сопровождается определёнными признаками.
Симптомы патологии
Чаще синдром Рихтера развивается на поздних стадиях и значительно осложняет течение хронического лимфолейкоза. Он проявляется:
- массивной лимфаденопатией;
- быстрым увеличением лимфоузла.
У некоторых пациентов по мере развития крупноклеточной диффузной лимфомы исчезает основной признак лимфолейкоза – увеличенное количество лимфоцитов в крови. У других же наоборот, лимфоцитоз значительно возрастает.
Помимо этого у больных проявляются симптомы опухолевой интоксикации и признаки ослабления гуморального иммунитета. Пациенты жалуются:
- на обильное ночью;
- интенсивное беспричинное ;
- повышенную температуру тела;
- частые ;
- постоянно возникающие ;
- длительно заживающие кожные раны.
Боль возникает там, где расположены поражённые лимфоузлы.
Осложняется течение заболевания, если в патологический процесс вовлекается костный мозг и другие органы кроветворения. Нередко при синдроме Рихтера врач выявляет . И тогда пациенты жалуются на боль, давящее ощущение в брюшной полости.
Чтобы поставить точный диагноз, необходимы , биопсия лимфоузла. Для гистологического исследования материал берут из наиболее подозрительных участков. Забор биоптата проводят с различных мест.
Прогноз при синдроме Рихтера
Дальнейшее прогрессирование болезни зависит от тактики лечения и индивидуальных особенностей пациентов. Часть пациентов после появления крупноклеточной лимфосаркомы умирают в течение полугода, даже если использовать адекватные для агрессивных лимфом комбинированные методы терапии.
Не всегда синдром Рихтера приводит к скорому летальному исходу. Особенно если самочувствие не изменяется. В этом случае продолжительность жизни при синдроме Рихтера значительно варьирует. Пациент может прожить от 3,5 месяцев до 9 лет.
При синдроме Рихтера пациенты нуждаются:
- в паллиативном лечении;
- спленэктомии (при значительно увеличенной селезёнке);
- комбинированном лечении.
При подборе схемы химеотерапии учитывают тип лимфом. Нередко цитостатики, цитотоксины дополняют противовоспалительными препаратами.
Пациентам с лимфомами, особенно при развитии синдрома Рихтера, необходимо избегать инфекционных заболеваний. Часто причиной смерти становится не сама опухоль, а неспособность иммунитета противостоять болезнетворным микроорганизмам.
К какому врачу обратиться
 Больным синдромом Рихтера важно предупреждать развитие инфекционных заболеваний, поскольку причиной смерти при таком диагнозе нередко становятся именно они.
Больным синдромом Рихтера важно предупреждать развитие инфекционных заболеваний, поскольку причиной смерти при таком диагнозе нередко становятся именно они. Синдром Рихтера развивается у пациентов с лимфопролиферативными заболеваниями. Такие больные находятся под наблюдением у врачей-гематологов, онкогематологов. Следует периодически проходить необходимые исследования, а при ухудшении самочувствия обязательно обратиться к лечащему врачу, чтобы вовремя выявить синдром Рихтера и изменить тактику лечения.
Течение заболевания носит аутоиммунный характер, иначе говоря, иммунная система человека начинает работать в обратном направлении, вырабатывая антитела, не защищающие больной орган, а поражающие его. Наиболее часто синдромом Рейтера страдают молодые мужчины вследствие неизлеченного хламидиоза, перешедшего в хроническую стадию.
Причины возникновения
Помимо заражения хламидиями вследствие незащищенного сексуального контакта, синдром Рейтера может развиться в результате перенесенного человеком энтероколита, вызванного сальмонеллой. Иммунная система дает сбой и начинает неадекватно реагировать на появление чужеродных тел. Организм вырабатывает антитела, направленные на поражение собственных тканей, помогая тем самым чужим антигенам. Таким образом, первыми начинают страдать суставные соединительные ткани, которые разрушаются под давлением иммунной системы.
В настоящее время неизвестно, чем обуславливается возникновение синдрома Рейтера у одних, и отсутствие у других. Современная медицина основной причиной называет расположенность к сбоям иммунной системы на генетическом уровне. Отчасти этим объясняется, почему болезнь Рейтера довольно часто является предвестником СПИДа: патологии иммунитета провоцируют мочеполовую или кишечную инфекцию вовлекать в болезнь соседние органы.
Что касается синдрома Рейтера в результате перенесенной половой инфекции, то больше всего она поражает мужской пол на пике сексуальной активности, в возрасте от 20 до 40 лет. Женщины болезнью суставов страдают нечасто, в основном, они являются носителями возбудителей хламидиоза.
А вот при кишечной инфекции в одинаковую зону риска попадают и мужчины, и женщины, и дети, причем у 80% заболевших – это результат генетической предрасположенности.
Особенности болезни
Синдром Рейтера развивается поэтапно, вовлекая болезненный процесс органы один за другим, не поражая все одновременно.
Начинается все с вышеописанной инфекции. Так как бывает, что недуг не дает видимых симптомов, то и связи с воспалением не находят. Но в основном анамнез болезни Рейтера достаточно четок, чтобы составить целую картину:
- Поражение мочеполовой системы или кишечника;
- Воспаление органов зрения;
- Воспаление суставных тканей.
Два первых симптома могут длиться недолгое время незамеченно. И только когда пациент приходит с жалобами на суставные боли, врач определяет связь в возникших за короткий промежуток времени недомоганий глаз, мочеполовой сферы и суставов.
Симптомы
Развитие болезни проходит в два этапа.
Инфекционный - в результате сексуального контакта хламидии попадают в мочеполовую сферу, где начинают распространяться (уретра, простата у мужчин, шейка матки у женщин). Этот процесс занимает от пары дней до месяца.
У мужчин проявляются такие симптомы:
- На половом органе возникает зуд и чувство жжения.
- Выход уретры краснеет и отекает.
- Появляются выделения.
- Мочеиспускание становится болезненным.
Болезнь перетекает в стадию хронической, и воспаление переносится на придатки и предстательную железу. Как итог – простатит и эпидидимит.
Симптомы заболевания у женщин следующие:
- В шейке матки развивается воспаление (цервицит);
- Возникают боли внизу живота;
- Появляются слабовыраженные гнойные выделения;
- При мочеиспускании появляется резь;
- Половой акт вызывает неприятные ощущения.
Хроническая стадия хламидиоза у женщин приносит с собой нарушение менструального цикла и кровотечения из матки.
Синдром Рейтера вследствие инфекции кишечника начинается сначала как обычное расстройство ЖКТ. В результате употребления продуктов ненадлежащего качества проявляются такие симптомы, как:
Вследствие интоксикации больной мучается ознобом, лихорадкой, ломотой суставов и головной болью.
Иммунопатологический - инфекция выходит за рамки мочеполовой сферы и начинает пожирать слизистую оболочку и суставные ткани организма.
- Слизистая век отекает.
- Выделения, как при конъюнктивите.
- Появляется зуд в глазных яблоках, синдром «попадания песка».
- Слизистая краснеет, на склере расширяются сосуды.
Иногда возникают патологические процессы в радужке, роговице, зрительном нерве.
Даже «запущенные» проблемы с суставами можно вылечить дома! Просто не забывайте раз в день мазать этим.
Основной признак синдрома Рейтера – сбой в функционировании опорно-двигательного аппарата.
Признаки заболевания суставов:
- Появляется боль в суставах ног (колени, голеностопы, фаланги пальцев).
- Пик болевых ощущений приходится на ночь и раннее утро.
- Кожный покров в области суставов краснеет, доходя до багрового оттенка на пальцах.
- Больные суставы начинают отекать до шарообразного состояния.
Болезнь Рейтера характерна тем, что поражает суставы ног по восходящей начиная от фаланг пальцев и поднимаясь до коленного сустава. При этом суставы верхних конечностей болезнь затрагивает очень редко, и то при полном отсутствии лечения болезни.
Важный симптом синдрома Рейтера – асимметрия воспалительных признаков. На левой ступне это могут быть, допустим, пальцы и плюсна, а на правой – пятка и ахиллесово сухожилие.
Помимо воспаления органов зрения, может наблюдаться язвенный стоматит во рту, на головке пениса появляются язвы.
Ступни и ладошки покрываются участками с ороговевшей шелушащейся кожей. Ногтевые пластины ломаются и желтеют.
При переходе болезни в тяжелую стадию поражаются важнейшие внутренние органы:
- Сердце – выражается миокардитом;
- Легкие – развивается плеврит или пневмония;
- Нервная система – возникает менингоэнцефалит или полинейропатия.
Диагностика заболевания
Сбор полной информации о развитии и течении болезни для постановки правильного диагноза крайне важен. И главным определяющим фактором является наличие урогенитальной инфекции. Ни в коем случае не стоит скрывать ее наличие из чувства ложного стыда. Только полная клиническая картина даст врачу возможность поставить правильный диагноз.
После составления анамнеза проводится ряд лабораторных исследований образцов крови, мочи, конъюнктивы и слизистых оболочек уретры у мужчин и шейки матки у женщин. Иногда на анализ берут семенную жидкость для выявления степени распространения инфекции.
Когда синдром Рейтера переходит в стадию иммунопатологии и идет поражение суставных тканей, требуется анализ синовиальной жидкости, взятой при помощи пункции. Такое исследование крайне важно, если течение болезни отягощается сердечными патологиями – оно позволяет точно установить причину возникновения артрита, выяснив, инфекция тому виной или ревматизм.
Кроме лабораторных исследований, проводят рентген суставов для определения суставных патологий, сопровождающих заболевание.
Посевом кала выявляют возбудителей инфекций кишечника. Анализом генов определяют наличие предрасположенности к патологиям ревматического характера.
Лечение
Болезнь Рейтера – в своем течении сложная, и заниматься ее лечением необходимо с привлечением специалистов самых разных направлений. Независимо, на какой стадии находится заболевание, тактику лечения вырабатывают только после консультации больного с рядом врачей: уролога, ревматолога, офтальмолога и других специалистов.
Делается это с целью исключить возникновение осложнений или рецидива после проведенного курса лечения.
Лечение медикаментами имеет две основополагающих: противовоспалительное и антибактериальное.
Для подавления инфекции используют 2-3 различных вида антибиотиков, которые принимают в диапазоне от 2 до 3 недель:
- Тетрациклин или Доксициклин.
- Ципрофлоксацин, Ломефлоксацин, Офлоксацин.
- Эритромицин, Азитромицин, Кларитромицин.
Противовоспалительная терапия проводится с целью устранения воспалительных течений в суставных тканях вследствие поражения хламидиями:
Необходимо менять лекарственный препарат по истечении двух недель лечения болезни, чтобы избежать его привыкания организмом.
Одновременно с этим проводится лечение осложнений. Сюда включено лечение гормонами, антигистаминами и другие виды терапии.
Крайне необходимо параллельно с лечением синдрома Рейтера купировать сопутствующие его заболевания – холецистит, простатит, ОРЗ и остальные заболевания, тормозящие эффективность лечения, замедляющие процесс выздоровления и способные вызвать осложнение.
Вспомогательные способы лечения синдрома Рейтера
После снятия острого воспаления в суставах, для наиболее эффективного лечения подключают физиотерапию:
- Фонофорез суставов с лекарственными аппликаторами;
- Магнитотерапия.
На стадии только начинающегося синдрома Рейтера применяют комплекс ЛФК, с целью сохранения подвижности суставов. При возникновении атрофии мышц применяют массаж и минеральные ванны с радоном или сероводородом.
Для лечения и профилактики БОЛЕЗНЕЙ СУСТАВОВ и ПОЗВОНОЧНИКА наши читатели используют метод быстрого и безоперационного лечения, рекомендованный ведущими ревматологами России, решившими выступить против фармацевтического беспредела и представивших лекарство, которое ДЕЙСТВИТЕЛЬНО ЛЕЧИТ! Мы ознакомились с данной методикой и решили предложить её вашему вниманию. Читать подробнее.
Как забыть о болях в суставах?
- Боли в суставах ограничивают Ваши движения и полноценную жизнь…
- Вас беспокоит дискомфорт, хруст и систематические боли…
- Возможно, Вы перепробовали кучу лекарств, кремов и мазей…
- Но судя по тому, что Вы читаете эти строки - не сильно они Вам помогли…
Хотите получить такое же лечение, спросите нас как?
ГЛАВА 25 СИНДРОМ РИХТЕРА
Синдром Рихтера представляет собой своеобразный и уникальный в своем роде клинико-морфологический феномен, характеризующийся сочетанием двух, обычно последовательно развивающихся злокачественных процессов - лимфоцитарного и крупно клеточного.
Впервые «генерализованная ретикулосаркома лимфатических узлов, ассоциированная с лимфатической лейкемией» была четко описана М.К.ШсЫег в 1928 г. . С тех пор в связи с кардинальным изменением представлений о гемо- поэзе и широким внедрением иммунологических методов исследования лимфоидная природа крупноклеточной опухоли, осложняющей течение хронического лимфолейкоза или лимфосаркомы с лимфоцитарным типом лейкемизацин, была доказана и в настоящее время не является предметом дискуссии. Тем не менее на протяжении 70 лет по существу остается открытым главный патогенетический вопрос, логически вытекающий из проблемы «ассоциированных» лимфопролиферативных заболеваний - о происхождении крупноклеточной лимфосаркомы: является ли она результатом трансформации лимфоцитов (пролимфоцитов) или это вторая опухоль, клонально (генетически) не связанная с лимфоцитарной лейкемиза- цией/хроническим лимфолейкозом?
Казалось бы, синдром Рихтера представляет собой довольно простую клиническую ситуацию, когда у больного последовательно обнаруживаются две различающиеся между собой, чаще В- клеточные опухоли - лимфоцитарная с лимфо- цитозом крови/костного мозга и крупноклеточная экстра медуллярная. Однако за кажущейся простотой явления скрывается чрезвычайно «проблемный» с точки зрения интерпретации его сущности клинико-морфологический феномен.
Приходится признать, что современный уровень знаний не позволяет сформулировать единую концепцию патогенеза «ассоциированных» лимфопролиферативных заболеваний. Главное достижение последних лет - переход от клеточного на молекулярно-генетический уровень обследования больных с синдромом Рихтера с изучением реаранжировок/мутаций генов иммуноглобулинов, некоторых онкогенов (р53 и т.д.) позволил только приблизиться к окончательному решению проблемы. Труднее всего доказать, что заболевания происходят из разных клонов и между онкогенными событиями, приведшими к развитию сначала лимфоцитарной, а затем крупноклеточной опухоли нет никакой генетической (клональной) связи. Иными словами, подтвердить одноклоновое происхождение лимфоцитарной лейкемизации/хронического лимфолейкоза и крупноклеточной лимфосаркомы при синдроме Рихтера значительно проще, чем его отвергнуть. Даже обнаружение различных типов перестройки генов иммуноглобулинов в клетках лимфоцитарной и крупноклеточной опухолей не всегда свидетельствует о происхождении этих заболеваний из разных клонов. По-видимому, это связано с самой природой иммуноглобулиновых генов, подверженных соматическим гипермутациям, де- лециям, переключению классов с селекцией наиболее аффинных клонов клеток.
Скорее всего понятие «синдром Рихтера» значительно шире, чем представляется на первый взгляд, и включает случаи как клональной прогрессии, так и возникновения вторых опухолей. В определенной степени это подтверждают результаты морфонммунологических и углубленных молекулярно-генетических исследований, которые при всей сложности проблемы все же проливают свет на клональные взаимоотношения между двумя злокачественными процессами - лимфоцитарным и крупноклеточным. Вместе с тем вопрос
о том, существует ли один клон или два неродственных, одна болезнь или две самостоятельные, часто остается открытым. По существу синдром Рихтера превратился в особую и чрезвычайно сложную область иммунологических и молекулярно-генетических исследований.
Известно, что далеко не всякая эрелоклеточ- ная лимфатическая опухоль заканчивается присоединением крупноклеточной лимфосаркомы. Бо
лее того, синдром Рихтера - это редкий клинико-морфологический феномен и, по данным различных авторов, встречается лишь у 3-10 % больных хроническим лимфолейкозом/лимфоци- тарной лимфомой .
Судя по литературным данным, развитие крупноклеточной лимфосаркомы у больных зрелоклеточными лимфопролиферативными заболеваниями является плохим в прогностическом отношении признаком и, как правило, сопровождается ухудшением состояния, появлением общих симптомов и генерализацией экстрамедуллярного опухолевого процесса . Продолжительность жизни после обнаружения крупноклеточной саркомы обычно не превышает полугода, несмотря на применение адекватных при лимфомах высокой степени злокачественности методов комбинированной химиотерапии .
Анализ 8 наших наблюдении синдрома Рихтера свидетельствует о том, что развитие крупноклеточной лимфомы по ходу течения хронической лимфатической опухоли не всегда означает терминальное состояние, более поздний этап опухолевой прогрессии и плохой прогноз. Только у половины больных подобная «трансформация» сопровождалась ухудшением состояния и присоединением общих симптомов, у остальных же самочувствие оставалось без изменений. После установления диагноза крупноклеточной лимфосаркомы продолжительность жизни варьировала от 3,5 мес до 9 лет.
При синдроме Рихтера иногда наблюдается изолированная экстранодальная локализация очагов крупноклеточной лимфосаркомы. Так, описано изолированное поражение стекловидного тела , кожи , мягких тканей с деструкцией позвонка , вещества мозга , яичек , желудка и(или) кишечника , почек , бронхиального дерева с эпдобронхиальным ростом опухоли .
Необходимо иметь в виду, что спектр злокачественных лимфопролиферативных заболеваний, протекающих с лимфоцитарной инфильтрацией костного мозга и лейкемической картиной крови, не ограничивается хроническим лимфолейкозом и его клиническими вариантами. При всех зрелоклеточных формах лимфом (лимфоцитарная, лим- фоппазмоцитарная, центрофолликулярная, ман- тийиоклеточная, из клеток маргинальной зоны, в том числе МАШ”) возможно раннее вовлечение в опухолевый процесс костного мозга с развитием картины хронического лимфолейкоза. Одним словом, под названием «лимфоцитарная лейкеми- зация» может скрываться широкий спектр зрелоклеточных лимфопролиферативных опухолей, при которых возможно развитие крупноклеточной лимфосаркомы, что полностью соответствует представлениям о синдроме Рихтера.
Название «крупноклеточная лимфома» тоже объединяет ряд лимфосарком с диффузным характером роста: центробластную (макролимфо- бластную), иммунобластную, из клеток с многодольчатыми ядрами, а также крупноклеточную а н а пла сти ч ескуто,
Особый интерес вызывают те редкие клинические наблюдения, в которых развитие крупноклеточной лимфосаркомы сопровождается исчезновением лимфоцитоза крови и костного мозга . Некоторые авторы описывают подобные случаи как редкий вариант синдрома Рихтера с регрессией хронической лимфатической лейкемии . Объяснить такое течение лимфоцитарной опухоли трудно. Не исключено, что именно в этих случаях речь может идти
о трансформации (клональной прогрессии) одного варианта злокачественной неходжкинской лимфомы в другой, более агрессивный.
Однако предположение о трансформации не находит должного морфологического подтверждения. Дело в том, что в тканевых инфильтратах чаще всего одновременно обнаруживаются опухолевые изменения двух отчетливо различающихся типов - лимфоцитарного и крупноклеточного. Создается впечатление, что обе опухоли сосуществуют и развиваются одновременно в одних и тех же тканях и органах. Проведенные нами исследования подтверждают сказанное. Действительно, в гистологических препаратах переходные между малыми лимфоцитами и крупными лимфоидными элементами клеточные формы не встречались. Однако при изучении отпечатков у отдельных больных цитограмма была смешанноклеточной: между самыми маленькими клетками - лимфоцитами и самыми крупными - нм- мунобластами, обнаруживалась довольно заметная популяция опухолевых элементов средней величины, которые, возможно, находились на различных этапах морфологической дифференцировки и которые вполне можно было отнести к переходным формам.
В период развития крупноклеточной (иммуно- бластиой) лимфосаркомы более чем у половины больных мы наблюдали самопроизвольную регрессию лимфоцитоза, т.е. исчезновение основного признака лимфоцитарной опухоли. Однако основу комбинированных лимфопролиферативных заболеваний составляют не столько случаи с регрессией лимфоцитоза (их чрезвычайно мало), сколько синдром Рихтера, когда обе болезни - лимфоцитарная и крупноклеточная - сосуществуют параллельно, часто поражая одни и те же ткани, в том числе костный мозг. Следовательно, исчезновение лимфоцитоза при развитии крупноклеточной опухоли - отнюдь не закономерность. Напротив, в наших наблюдениях у 2 больных генерализация иммунобластной лимфомы сопро
вождалась ростом лимфоцитоза крови и костного мозга до самых высоких значений за весь период наблюдения.
Иммунологическое изучение клеток злокачественной лимфомы на современном уровне предполагает не только оценку линейной принадлежности, состояния активации или покоя, но и определения степени дифференцировки. Обнаружение диффузной крупноклеточной лимфомы при картине крови и костного мозга, характерной для лимфоцитарной опухоли, только на первый взгляд представляет сложную для интерпретации клиническую ситуацию, В таких, случаях для правильной диагностики, оптимального решения тактических вопросов и выбора наиболее адекватного метода химиотерапии очень важно располагать в том числе и результатами нммуноло-
1. Аникин Б.С.. Лихачев А.А.. Пекина Л.Н. и др. Тер. арх.-1979.-№ 9,-С.118-121.
2. Арутюнов ВД. Арх. пат,-1956,-№ I.-С.56-59.
3. Демидова А.В,. Воробьев А.И., Даценко С.Ф. и др. Пробл. гематол.-1967.--Т.!2, № И.-С.10-17.
4. Крьтое А.А. Тер. арх.-1974.-№ 8,-С.49-51.
5. Пробатова НА., Мамедов Р.Д., Круглова Г.В. Арх. пат,-1988.-Т.Ь, № 3.-С,37-43.
6. Файя\итейн Ф.З., Полянская А М. Тер. арх.-1984.-№ 10.-С-80-83.
7. АШог С.. ()1.-1987,-УоШ, N 12.-Р.901-903.
13.Оипп Р., Кии Т.Т.. Тгеп Н.Г. Ь Рогггюз МесЬ А 85-
1995. -Уо1.94, N 1Ь-Р.686-688.
14.Ш%е1 О.О., Усш§кп О.У., ЕипН Е.Е. ег а1. Атег. Ь Саз1гоепС,-!995,-Уо1.90, N 4.-Р.635-637.
15.Еогет V., Соп/Ыотеп Р. Мтшуа Мей-1984.-Уо1.75, N 45-46.-Р.2741-2749.
16.Ежсат К., Нут ЯЕ Сапсег -1980.-’Уо1.4«.-РЛ18-134.
Болезнь Рейтера
Болезнь Рейтера (синдром Рейтера) - развивающееся последовательно или одновременно сочетанное поражение глаз (конъюнктивит), суставов (реактивный моно- или олигоартрит) и мочеполовых органов (чаще неспецифический уретропростатит). Болезнь Рейтера развивается при условии наличия у генетически предрасположенных людей хламидийной инфекции. Как правило данная болезнь развивается в молодом возрасте, мужчины болеют чаще женщин приблизительно в двадцать раз. Пик заболеваемости приходится на возрастной промежуток от двадцати до сорока лет (порядка 80%). Встречаются единичные случаи заболеваемости детей
Болезнь Рейтера - причины возникновения
Наиболее часто в развитии болезни Рейтера помимо генетической предрасположенности играют роль различные инфекции пищеварительной и мочевыводящей систем. Обычно заболевание начинается с уретрита, который возникает после обострения какой-либо хронической инфекции мочевыводящих путей или после полового контакта. При наличии неблагоприятной эпидемической обстановки, которая зачастую присутствует в туристических и военных лагерях, катализатором развития болезни Рейтера может быть острый энтероколит иерсиниозного, сальмонеллезного или шигеллезного происхождения. В механизме поражения суставов основное значение придают наследственной предрасположенности и иммунным процессам.
Болезнь Рейтера может быть вызвана некоторыми типами хламидий, которые обычно поражают слизистые оболочки человека, проникая в его организм через мочеполовые органы и поражая в последствие другие системы и органы. Вследствие того, что хламидии могут достаточно длительно находиться в организме больного, велика вероятность появления обострений и рецидивов данного заболевания, или развитию хронической болезни Рейтера
На первый план в клинической картине болезни Рейера выходят проявления конъюнктивита, артрита и уретрита. Помимо этого могут наблюдаться изменения слизистых оболочек и паренхиматозных органов (ЦНС, аорты, миокарда, почек, печени и пр.).
Последовательность основной симптоматики может быть различной, однако наиболее часто болезнь Рейтера начинается с развития таких болезней как цистит, уретрит или простатит. При данном заболевании уретрит может быть разным по своей выраженности - от стертого, склонного к затяжному/хроническому течению, до острого с наличие сильных гнойных выделений. Уретрит проявляется жжение, зудом, скудными выделениями из влагалища и уретры, гиперемией вокруг выходящего отверстия мочеиспускательного канала и малоприятными ощущениями при мочеиспускании. У выделений обычно слизистый характер.
Вскоре после уретрита у человека наступает поражение глаз, которое наиболее часто проявляется конъюнктивитом, реже кератитом, ретробульбарным невритом, ретинитом, увеитом, иридоциклитом, иритом. Конъюнктивит при болезни Рейтера чаще всего бывает двусторонним, слабовыраженным и проходящим по истечении одного-двух дней. Очень часто он остается незамеченным.
Поражение суставов при болезни Рейтера является ведущим признаком и развивается по истечении одного-полутора месяцев с момента развития острой мочеполовой инфекции. Наиболее характерным поражением является асимметричный артрит, вовлекающий в процесс суставы нижних конечностей - межфаланговые, плюснефаланговые, голеностопные и коленные. Боли в суставах обычно усиливаются утром и ночью, кожные покровы над ними гиперемированы, присутствует выпот. По истечении нескольких дней наблюдается характерное последовательное (снизу вверх) вовлечение суставов. Нередко наблюдается развитие пяточных шпор, пяточный бурсит, воспаление ахиллова сухожилия. У некоторых пациентов наблюдаются боли в позвоночнике, что в последствие приводит к развития саркоилеита.
У 30% больных отмечаются рецидивы артрита; у 20% больных артрит переходит в хроническую стадию с атрофией прилежащих мышц, а также ограничением функции суставов; у 50% больных суставная симптоматика полностью исчезает. Вследствие поражения суставов предплюсны у некоторых больных может развиться плоскостопие. Поражение суставов верхних конечностей наблюдается крайне редко.Порядка 50% больных подвержены поражению кожных покровов и слизистых оболочек. В районе головки полового члена и на слизистой полости рта появляются болезненные язвы, развивается баланопостит или баланит, возможно развитие глоссита и стоматита. Поражение кожных покровов характеризуется появлением небольших по размеру красных папул, а иногда эритематозных пятен. Для болезни Рейтера характерна кератодермия, которая на фоне гиперемии кожи выражается сливными очагами гиперкератоза с шелушением области ладоней и стоп. Также зачастую очаги гиперкератоза наблюдаются на кожных покровах туловища и лба.
При синдроме (болезни) Рейтера может наблюдаться безболезненное увеличение лимфатических узлов (чаще паховых); у порядка 20% больных присутствуют признаки поражения сердца (миокардит, миокардиодистрофия), поражение почек (амилоидоз почек, нефрит), нервной системы (полиневрит), легких (плеврит, очаговая пневмония) и длительная субфебрильная температура тела
Диагностика болезни Рейтера
При наличии характерной для данного заболевания триады (конъюнктивит+уретрит+артрит), диагностика болезни Рейтера не вызывает практически никаких затруднений. При недостаточной выраженности отдельной симптоматики, или в атипичных случаях, показано проведение рентгенологического исследования суставов. Проведение анализа синовиальной жидкости выявит признаки воспаления. Биопсия синовиальной жидкости позволит выявить картину неспецифического подострого или острого воспаления. Общий биохимический анализ крови отклонений не выявляет. В моче наблюдается появление гноя
Лечение болезни Рейтера исключительно по поводу поражения суставов, которое является наиболее беспокоящим и ярким проявлением болезни, не дает желаемых результатов и обычно приводит к затяжному или хроническому течению заболевания. К такому же результату приводит лечение препаратами цефалоспориновой и пенициллиновой группы. Необходимо проводить лечение как самого больного, так и его полового партнера.
При болезни Рейтера все лечебные мероприятия можно условно подразделить на такие основные направления как противовоспалительная терапия суставного вещества и антибактериальная терапия инфекции.
Антибактериальное лечение уретрита прежде всего проводится препаратами тетрациклинового ряда. В лечении артрита применяют такие нестероидные противовоспалительные препараты как вольтарен, индометацин, аспирин, диклофенак и пр. В случае высокой активности заболевания и при ярко выраженных системных проявлениях, показано применение глюкокортикоидов. В случае хронического или затяжного артрита показано применение солей золота или производных хинолина.
Первичная профилактика болезни Рейтера основана на соблюдении стандартных санитарно-гигиенических правил, своевременном лечении цистита, уретрита и прочих мочеполовых инфекций. В случае наличия хламидийной инфекции, обязательно лечение обоих половых партнеров.
Синдром Рейтера: симптомы и лечение
Синдром Рейтера сопровождается триадой воспалительных поражений суставов, глаз и мочеполовых органов. В 80 % случаев он наблюдается у молодых мужчинлет, реже – у женщин, крайне редко – у детей. При отсутствии лечения может вызвать тяжелые осложнения – вплоть до инвалидизации больного.
В этой статье мы ознакомим вас с симптомами и основными способами лечения и профилактики синдрома Рейтера. Владея этой информацией, вы сможете вовремя принять решение о необходимости обращения к врачу для предотвращения таких осложнений этого патологического процесса, как хронизация заболевания, нарушение подвижности позвоночника и развитие ухудшения зрения (вплоть до слепоты).
Впервые синдром Рейтера был описан как осложнение кишечной инфекции, а позднее стало известно, что он может провоцироваться и инфекционными процессами в мочеполовой системе. Причиной развития этого недуга становится аутоиммунная реакция, возникающая в ответ на внедрение бактериального или вирусного агента.
Чаще он развивается на фоне хламидиоза, а иногда выявить его точную причину развития не удается.
Кроме хламидий синдром может провоцироваться уреаплазмами, сальмонеллами, шигеллами и иерсиниями. И большинство специалистов склоняются к теории о наличии наследственной предрасположенности к возникновению такой аутоиммунной реакции в ответ на инфекцию.
Симптомы
Синдром Рейтера развивается спустя 1,5-2 месяца после перенесенной мочеполовой или кишечной инфекции. А его течение может быть:
- острым – до полугода;
- затяжным – до 1 года;
- хроническим – более 1 года.
Симптомы со стороны мочеполовой системы
Именно признаки поражения мочеполовой системы чаще становятся первыми сигналами о начале развития синдрома Рейтера. Они проявляются симптомами уретрита, цистита, простатита, вагинита и пр.
У мужчин обычно появляются такие симптомы:
- неприятные ощущения во время опорожнения мочевого пузыря: зуд, жжение, слизистые выделения;
- учащенное мочеиспускание;
- гиперемия наружного отверстия уретры;
- боли или дискомфортные ощущения во время секса;
- боли внизу живота.
У женщин обычно появляются такие симптомы:
- выделения из влагалища;
- жжение, боли и рези при мочеиспускании;
- учащенное мочеиспускание;
- боли во время полового акта;
- дискомфортные ощущения или боль внизу живота.
В лабораторных анализах – мазках и моче – определяется лейкоцитоз.
Симптомы со стороны органов зрения
Через короткий промежуток после появления признаков поражения мочеполового тракта у больного появляются признаки воспаления глаз. Впоследствии они приводят к развитию конъюнктивита, а в тяжелых случаях вызывают ирит, иридоциклит, ретробульбарный неврит, увеит или кератит.
При синдроме Рейтера больного беспокоят следующие симптомы поражения глаз:
- боль и дискомфортные ощущения;
- слезотечение;
- слизистые или гнойные выделения;
- ухудшение зрения;
- покраснение глаз;
- светобоязнь.
Иногда слабовыраженные проявления конъюнктивита наблюдаются только на протяжении первых двух дней и остаются незамеченными.
Симптомы со стороны суставов
Основным проявлением синдрома Рейтера является поражение суставов, которое впервые дает о себе знать спустя 1-1,5 месяцев после появления признаков поражения мочеполовой системы или их обострения. Обычно при этом заболевании происходит воспаление 1-2 суставов (моно- или олигоартрит), но иногда течение патологического процесса захватывает множество суставов и у больного развивается полиартрит. Чаще воспаляются суставы ног и этот процесс распространяется по принципу снизу-вверх (т. е. вначале развивается артрит голеностопного сустава, а затем – коленного и т. п.).
При синдроме Рейтера больного беспокоят следующие симптомы поражения суставов:
- боль;
- асимметричность поражения суставов;
- изменение окраски кожи над суставом (от красного до синюшного цвета);
- гипертермия и отечность кожных покровов в области воспаления.
В некоторых случаях при синдроме Рейтера происходит поражение крестцово-подвздошного сочленения и суставов позвоночного столба. При этом у больного появляется скованность в движениях по утрам и боль. А при поражении суставов стопы может происходить быстрое формирование плоскостопия.
По данным статистики у половины больных симптомы артритов исчезают полностью, у 30 % – возникает рецидивирующий артрит, а у 20 % – хронический артрит, приводящий к ограничению функциональности суставов и атрофии прилегающих мышц.
Другие симптомы
Иногда при синдроме Рейтера, всегда сопровождающемся триадой характерных симптомов, появляются признаки поражения других органов.
На коже могут появляться красные пятна, которые возвышаются над ее поверхностью в виде бугорков. Как правило, такие изменения наблюдаются на ладонях и подошвах. В дальнейшем возможно образование уплотненных зон с признаками шелушения и ороговения кожных покровов.
Иногда при синдроме происходит поражение слизистых оболочек. Такие признаки наблюдаются на половых органах и в полости рта.
На фоне артритов могут возникать воспалительные процессы в области крепления сухожилий и связок. Такие процессы сопровождаются появлением боли, покраснений и отечности. Как правило, такой воспалительный процесс локализуется в области ахиллова сухожилия.
В крайне редких случаях синдром Рейтера приводит к воспалительным процессам в почках, легких или сердце.
Диагностика
Предположительный диагноз «синдром Рейтера» может ставиться на основании сведений о перенесенной мочеполовой или кишечной инфекции и присутствии в жалобах пациента данных о типичной для этой болезни триады симптомов. Для подтверждения диагноза больному назначается ряд лабораторных анализов:
- клинический анализ крови – лейкоцитоз, повышение СОЭ;
- соскоб из уретры или влагалища – выявление хламидий или уреаплазмы;
- анализ суставной жидкости – выявление хламидий;
- биохимия крови – отсутствие ревматоидного фактора и присутствие С-реактивного белка;
- соскоб слизистой глаза – выявление хламидий;
- иммунологический анализ крови – высокий титр иммуноглобулинов М и G;
- генетический анализ – определение гена HLA-B27;
- ПЦР крови – выявление ДНК хламидий/уреаплазм.
Для выявления нарушений в суставах и прилегающих к ним тканях могут назначаться такие инструментальные методы:
Лечение
Терапия синдрома Рейтера всегда комплексная и занимает от 3 до 12 месяцев. Ее основные цели направлены на устранение инфекционного агента, купирование воспалительного процесса и подавление аутоиммунной реакции.
Для лечения хламидиоза или уреаплазмоза назначают прием нескольких антибиотиков в максимальных дозах. Для предупреждения повторного инфицирования прием таких же препаратов рекомендуется половому партнеру. Больному могут назначаться комбинации следующих средств:
- макролиды: Клацид, Зи-фактор, Кларитромицин, Рокситромицин;
- фторхинолоны: Ципрофлоксацин, Спарфлоксацин, Офлоксацин;
- тетрациклины: Доксициклин.
Антибиотикотерапия проводится длительно – на протяжении 3-8 недель – и может приводить к развитию кандидоза и поражению органов пищеварительного тракта. Для предупреждения этих нежелательных последствий применяются следующие препараты:
- гепатопротекторы: Легалон, Гептрал, Карсил, Гепа-Мерц, Эссенциале, Гепабене и др.;
- противомикотические средства: Клотримазол, Пимафуцин, Флуконазол и др.;
- поливитаминные комплексы: Биовиталь, Алфавит, Дексавит, Витрум и др.
Для максимальной эффективности антибактериальной терапии рекомендуется параллельный прием протеолитических ферментов: трипсина, химотрипсина или вобэнзима.
Для лечения воспалительных поражений глаз применяются антибактериальные и противовоспалительные капли и мази на основе тетрациклина и эритромицина. Снизить воспалительные реакции позволяют примочки из настоев лекарственных трав (ромашка, календула и др.).
- нестероидные противовоспалительные: Нимесулид (или Нимегезик), Аркоксиа, Диклоберл, Целекоксиб;
- глюкокортикостероиды: Преднизолон, Полькортолон, Дипроспан, Кеналог.
Эти препараты позволяют устранить воспаление, боль, отечность и снижают температуру тела.
Важной частью лечения синдрома Рейтера является использование средств для подавления аутоиммунной реакции, направленной на разрушение соединительной ткани. Они применяются на протяжении длительного времени (4-12 месяцев), а в тяжелых случаях назначаются больному для пожизненного приема.
Для лечения синдрома Рейтера используются следующие иммуносупрессоры:
На фоне приема таких препаратов происходит снижение устойчивости организма к инфекционным заболеваниям и для их профилактики больному рекомендуется прием иммуномодуляторов:
Для повышения иммунитета могут использоваться такие методики, как ультрафиолетовое облучение крови и внутривенная квантовая терапия.
При повышении температуры и интоксикации больному назначаются десенсибилизирующие средства (Фенирамин, Лоратадин, Кетотифен) и внутривенное введение растворов реополиглюкина или реосорбилакта. Такая дезинтоксикационная терапия не только облегчает состояние больного, но и повышает эффективность других лекарственных средств.
После стихания воспалительного процесса назначается физиотерапия:
- лечебная физкультура;
- амплипульстерапия;
- магнитотерапия;
- электрофорез с раствором новокаина.
Профилактика
Специфических мер профилактики синдрома Рейтера не существует. Для предупреждения его развития рекомендуются меры, направленные на профилактику и своевременное лечение венерических заболеваний.
К какому врачу обратиться
Тяжесть синдрома Рейтера определяется поражением суставов, поэтому основную терапию назначает ревматолог. При сопутствующей патологии наружных мочеполовых органов необходима консультация уролога, гинеколога и венеролога. Поражение глаз - повод для консультации офтальмолога. Также необходимо лечение у физиотерапевта.
Специалист клиники «Московский доктор» рассказывает о синдроме Рейтера:
Первый канал, программа «Жить здорово!» с Еленой Малышевой, в рубрике «Про медицину» разговор о синдроме Рейтера (с 32:45):
Помоги детям
Полезная информация
Обратитесь к специалистам
Телефон службы записи к врачам-специалистам Москвы:
Информация предоставляется с целью ознакомления. Не занимайтесь самолечением. При первых признаках заболевания обратитесь к врачу.
Адрес редакции:, г. Москва, 3-я Фрунзенская ул., 26
2477 0
На основании клинико-лабораторной картины хронического лимфолейкоза (ХЛЛ) можно выделить два основных клинических синдрома и три основных лабораторных изменения в клиническом анализе крови, доступных выполнению в любом лечебном заведении.
Клинические синдромы это:
1) генерализованная лимфаденопатия: лимфоузлы мягко- или плотноэластической консистенции, безболезненные, не спаянные с окружающими тканями (за исключением случаев злокачественной трансформации), могут образовывать пакеты, кожа над ними не изменена, свищей не образуют;
2) гепато- и спленомегалия. Изменения в клиническом анализе крови: лейкоцитоз, абсолютный лимфоцитоз, клетки лейколиза (тени Боткина-Гумпрехта).
У многих пациентов долгие годы может отмечаться лишь лимфоцитоз - 40-50%, хотя общее количество лейкоцитов колеблется около верхнего предела нормы. Лимфатические узлы могут быть почти нормальных размеров, но они увеличиваются при различных инфекциях (например, при ангине), а после ликвидации воспалительного процесса сокращаются до исходной величины.
Лимфатические узлы увеличиваются постепенно, обычно в первую очередь на шее, в подмышечных впадинах, затем процесс распространяется на средостение, брюшную полость, паховую область. Возникают общие для всех лейкозов неспецифические явления: повешенная утомляемость, слабость, потливость. На ранних этапах болезни в большинстве случаев анемии и тромбоцитопении нет.
Отличительным признаком ХЛЛ является увеличение количества лейкоцитов периферической крови со значительным количеством малых зрелых лимфоцитов - более 5х10 9 /л (достоверным диагностическим признаком является их количество более 10х10 9 /л), выявление «теней Боткина-Гумпрехта» (клеток лейколиза) - разрушенных при приготовлении мазка лимфоцитов и наличие характерного иммунофенотипа лимфоидных клеток - CD5+, CD19+, Cd20+, CD22+, CD79a+, CD23+, CD43+, CD11c+/-, CD10-, циклин D1-.
Лимфоцитоз в крови постепенно нарастает: 80-90% лимфоцитов, как правило, наблюдается при почти тотальном замещении ими костного мозга. Распространение лимфатической ткани в костном мозге может годами не угнетать продукцию нормальных клеток. Даже при достижении высоких цифр лейкоцитов в крови 100000 в 1 мкл и более - анемии часто нет, количество тромбоцитов нормальное или незначительно снижено.
Пунктат костного мозга показывает увеличение процента лимфоцитов в миелограмме - обычно более 30. Этот признак надежен для диагностики хронического лимфолейкоза, если пунктат значительно не разбавлен периферической кровью. В трепанобиоптате отмечаются характерные разрастания лимфоидных клеток, чаще диффузные.
Морфология лимфоцитов при хроническом лимфолейкозе не имеет стабильных и типичных признаков. Она может меняться в течении болезни под влиянием вирусных инфекций. В крови большинство клеток составляют зрелые В-лимфоциты , ничем не отличающееся от нормальных.
Наряду с такими клетками могут быть лимфоцитарные элементы с более гомогенным ядром, не имеющие еще грубой глыбчатости хроматина зрелого лимфоцита, с широким ободком цитоплазмы, которая иногда, как при инфекционном мононуклеозе, имеет перинуклеарное просветление. Ядра клеток могут иметь своеобразную скрученность хроматиновых петель или быть правильно круглыми; встречаются и бобовидные ядра; цитоплазма бывает с обрывчатыми контурами, иногда с элементами «волосатости», но без гистохимических особенностей волосатоклеточного лейкоза.
Характерный признак хронического лимфолейкоза - полуразрушенные ядра лимфоцитов - тени Боткина-Гумпрехта. Их количество не является показателем тяжести процесса. Клетки лейколиза представляют собой артефакт: в жидкой крови их нет, они образуются в процессе приготовления мазка.
В начале болезни пролимфоцитов в лейкоцитарной формуле обычно нет. Однако бывают случаи хронического лимфолейкоза, которые с самого начала сопровождаются резким преобладанием в крови пролимфоцитов - клеток с гомогенным ядерным хроматином, но с отчетливой нуклеолой. На этом основании выделяют пролимфоцитарную форму хронического лимфолейкоза (Воробьев А.И. и соавт., 1985-2000). Иногда такой лейкоз может протекать с секрецией моноклонального иммуноглобулина (впрочем, это не составляет большой редкости и при обычном зрелоклеточном хроническом лимфолейкозе).
По мере развития болезни в крови начинают встречаться единичные пролимфоциты и реже лимфобласты. Их большое количество появляется лишь в терминальной стадии болезни.
Начальная стадия
Начало доброкачественного и прогрессирующего вариантов типичной формы ХЛЛ практически одинаково. В начальном периоде больные обычно не предъявляют значительных жалоб, общее состояние удовлетворительное. Однако некоторые пациенты даже в этом периоде могут жаловаться на небольшую слабость, потливость, частые простудные заболевания.Как правило, гемобластоз выявляется случайно (при профилактических осмотрах, при обращении к врачу по поводу какого-либо другого заболевания). Основными клиническими признаками ХЛЛ являются на этой стадии увеличение лимфатических узлов, лейкоцитоз и лимфоцитоз.
Наиболее часто в этой стадии отмечается небольшое увеличение лимфатических узлов, как правило, в определенной последовательности. Обычно в первую очередь увеличиваются шейные, затем подмышечные, а значительно позже (чаще всего в развернутой фазе заболевания) - другие группы лимфатических узлов. Увеличенные лимфоузлы при хроническом лимфолейкозе мягко-эластической тестоватой консистенции.
Следует подчеркнуть, что плотность лимфоузлов не характерна для этой стадии хронического лимфолейкоза. Размеры увеличенных лимфатических узлов различны: от небольшого до весьма значительного увеличения. Как правило, лимфатические узлы безболезненны, не спаяны с кожей и между собой, не изъязвляются и не нагнаиваются.
Второй характерный признак хронического лейкоза в начальном периоде - лейкоцитоз (обычно 10-30х10 9 /л) и увеличение количества лимфоцитов до 60-80%. А.И. Воробьев и соавт. (2003) указывает, что количество лейкоцитов в начальной стадии может повышаться до 50х10 9 /л.
Основными критериями начального периода хронического лимфолейкоза являются:
Незначительное или умеренное увеличение нескольких лимфатических узлов одной или нескольких групп;
лейкоцитоз, не превышающий 50х10 9 /л;
отсутствие тенденции к значительному увеличению лейкоцитоза;
удовлетворительное состояние больного, отсутствие нарушения функции других органов и систем (состояние компенсации).
При доброкачественном течении заболевания начальный период может продолжаться несколько лет. Нарастание лейкоцитоза медленное (в течение 2-3 лет). При развитии инфекционно-воспалительных процессов количество лейкоцитов и лимфоцитов в крови может значительно возрастать, но после купирования инфекции лейкоцитоз и лимфоцитоз возвращаются к прежним цифрам. Лимфоузлы и селезенка нормальных размеров или незначительно увеличены, консистенция узлов эластичная, размеры их не меняются.
При прогрессирующем течении хронического лимфолейкоза начальный этап заболевания продолжается недолго, количество лейкоцитов и лимфоцитов неуклонно нарастает из месяца в месяц, ухудшается общее состояние, отмечается значительное увеличение лимфатических узлов. Первыми обычно увеличиваются шейные, надключичные лимфоузлы, затем подмышечные, консистенция их тестоватая. Селезенка вначале не пальпируется или незначительно увеличена, в дальнейшем размеры ее значительно увеличиваются.
Период выраженных клинических проявлений (развернутая стадия)
В этом периоде имеет место развернутая клиническая картина хронического лимфолейкоза. Больные жалуются на выраженную общую слабость, снижение работоспособности, значительную потливость, особенно ночью, похудание, повышение температуры тела, увеличение лимфатических узлов.При осмотре обращает на себя внимание генерализованная лимфаденопатия. В этом периоде заболевания обычно увеличены практически все группы периферических лимфоузлов: подчелюстные, задние и передние шейные, надключичные, подмышечные, паховые и др. Степень увеличения лимфоузлов различна - от величины горошины до куриного яйца.
Консистенция лимфоузлов по-прежнему остается эластично-тестоватой, они не спаяны между собой и кожей. Однако при значительном увеличении лимфоузлов одной группы они могут выглядеть в виде конгломерата. С помощью специальных методов исследования (ультразвуковое исследование (УЗИ) , компьютерная томография (КТ) , рентгенография) обнаруживается также увеличение внутригрудных, внутрибрюшных и забрюшинных лимфатических узлов, однако при типичной (классической) форме ХЛЛ признаков компрессии внутренних органов не наблюдается в отличие от опухолевидной формы. В крови отмечается различной степени выраженности лейкоцитоз и абсолютный лимфоцитоз.
Академик А.И. Воробьев и соавт. (1985-2000) на основании морфологических и клинических признаков, включающих в том числе, и ответ на терапию, выделяют следующие клинико-лабораторные формы ХЛЛ:
1) Доброкачественная;
2) Прогрессирующая (классическая);
3) Опухолевая;
4) Спленомегалическая (селезеночная);
5) Абдоминальная;
6) Пролимфоцитарная;
7) Костномозговая.
Доброкачественная форма
По данным А.И Воробьева и соавт. (1985-2003), при доброкачественной форме ХЛЛ «отмечается очень медленное, заметное лишь на протяжении лет, но не месяцев, нарастание лимфоцитоза в крови параллельно с ростом числа лейкоцитов». Лейкоцитоз при этой форме ХЛЛ не высокий, как правило, менее 30х10 9 /л, в ряде случаев может достигать 50х10 9 /л. Процент лимфоцитов в крови составляет 60-70.Лейкоцитоз стабильно сохраняется ниже этого уровня не менее 3 лет от первого анализа крови с лейкоцитозом. Лимфатические узлы либо не увеличены, либо шейные увеличены весьма незначительно (не более 2 см). Очень медленное нарастание лейкоцитоза и лимфоцитоза до заметного увеличения лимфатических узлов может продолжаться и годы, и десятилетия.
Все это время больные находятся под диспансерным наблюдением, они полностью трудоспособны, им только запрещают повышенную инсоляцию (загорать нельзя, но можно купаться и отдыхать на юге, кроме июля и августа). Исследования крови с подсчетом тромбоцитов и ретикулоцитов делают каждые 1-3 месяца.
При доброкачественной форме до того момента, когда ухудшение состояния может потребовать терапии, во многих случаях не делают диагностической стернальной пункции, трепанобиопсии и биопсии лимфатического узла. Эти исследования существенно травмируют психику больного, которому зачастую до конца дней не потребуется цитостатических препаратов, но не могут ничего добавить к диагностике этой формы болезни. Правда, принимать подобные решения может лишь опытный специалист.
При данной форме, которую И.А. Кассирский называл «застывшей» продолжительность жизни вообще может не зависеть от наличия хронического лимфолейкоза, и пациенты пожилого возраста погибают от сопутствующей патологии, при отсутствии признаков прогрессии гемобластоза.
В доброкачественной форме заболевание протекает у 20-30% больных (Воробьев А.И. и соавт., 2003). Продолжительность жизни таких пациентов такая же, как и в популяции.
Прогрессирующая (классическая) форма
Начинается так же, как и доброкачественная, но количество лейкоцитов нарастает от месяца к месяцу, как и величина лимфатических узлов. Ведущим проявлением этой формы ХЛЛ служит значительное увеличение количества лейкоцитов. Лейкоцитоз может достигать 500-1000х10 9 /л и более.Одновременно в лейкоцитарной формуле увеличивается и количество лимфоцитов (до 90-99%). Преобладают зрелые формы, но, как правило, обнаруживается 5-10% пролимфоцитов. Содержание эритроцитов, гемоглобина и тромбоцитов вначале нормальное, а при высоком лейкоцитозе и значительном лимфоцитозе обычно снижены за счет вытеснения здоровых ростков патологическими лимфоцитами либо в связи с присоединением аутоиммунных осложнений.
Одновременно увеличиваются и размеры лимфатических узлов. Консистенция узлов может быть тестоватой, мягкой или слегка эластичной. «Деревянной» плотности обычно не бывает, а если такие узлы появляются, то их следует биопсировать для исключения злокачественной трансформации. В большинстве случаев незначительное увеличение лимфатических узлов можно обнаружить даже при нерезко выраженных изменениях в крови. Иногда первым симптомом их увеличения является реакция на инфекцию: при острых респираторных заболеваниях лимфоузлы увеличиваются, а затем снова уменьшаются в размерах.
Увеличение селезенки у таких пациентов в большинстве случаев появляется позже, чем увеличение лимфатических узлов и редко достигает значительных размеров. Еще позднее обычно увеличивается печень. Не отмечается высокой корреляции между степенью лимфоидной инфильтрации костного мозга, высоты лейкоцитоза и размеров лимфатических узлов, селезенки и печени.
Иногда в момент острого респираторного заболевания у пациента отмечается снижение слуха и «чувство заложенности» в ушах. При осмотре обнаруживается разрастание лимфоидной ткани у устьев евстахиевых труб, набухлость ее в период присоединения инфекции вызывает закрытие просвета труб.
В ряде случаев ХЛЛ клинические проявления заболевания (увеличение лимфатических узлов, селезенки, печени) отсутствуют у больных даже с очень высоким лейкоцитозом и лимфоцитозом. В подобных наблюдениях исследование костномозгового пунктата обычно выявляет почти тотальное вытеснение гранулоцитарных и эритроидных элементов костного мозга лимфоцитами.
Термин «классическая форма» означает, что в такой форме протекает большинство случаев ХЛЛ - 45-50% (Воробьев А.И. и соавт., 2003). Цитостатическая терапия этим больным обычно назначается при заметном нарастании всех проявлений болезни, лейкоцитоза (как правило, более 100-150х10 9 /л) и размеров лимфатических узлов в первую очередь. Медиана выживаемости составляет 96 месяцев (Никитин Е.А., Лорие Ю.Ю., Меликян А.Л., 2003).
Опухолевая форма
Особенностью этой формы, определившей ее название, является значительное увеличение и плотная конситстенция лимфатических узлов при невысоком лейкоцитозе. Миндалины увеличены, часто они почти смыкаются друг с другом. Увеличение селезенки обычно умеренное, но бывает и значительным.В лейкоцитарной формуле сохраняется достаточный - 20 и более - процент нейтрофилов. В костном мозге обычно не более 20-40% лимфоцитов, хотя бывает и тотальное его поражение. Несмотря на значительную гиперплазию лимфатической ткани, интоксикация долго мало выражена в отличие от генерализованной лимфомы, с которой иногда отождествляют эту форму хронического лимфолейкоза.
Суммируя все клинические, лабораторные и инструментальные данные можно выделить следующие особенности этой формы ХЛЛ:
Выраженное увеличение лимфатических узлов, они безболезненны, плотноэластической консистенции, сливаются между собой, образуют конгломераты. Сначала резко увеличиваются шейные, затем подмышечные и паховые лимфоузлы. Увеличение лимфоузлов можно диагностировать не только пальпаторно, конгломераты лифоузлов четко визуализируются. Часто первой жалобой таких пациентов является «невозможность застегнуть ворот рубашки», со слов пациентов «шея становится толстой», т.н. «борцовская шея». Наряду с увеличение периферических лимфоузлов возможно увеличение паратрахеальных лимфатических узлов со сдавлением трахеи и крупных бронхов. У некоторых больных увеличиваются внутрибрюшные лимфатические узлы со сдавлением воротной вены и желчевыводящих путей, что приводит к развитию синдрома портальной гипертензии и механической желтухи. Возможно увеличение ретроперитонеальных лимфоузлов со сдавлением мочеточников и нарушением оттока мочи.
Наличие в биптатах лимфоузлов диффузной пролиферации однородных лимфоидных клеток со светлыми ядрами; в отличие от лимфомы, отсутствуют признаки атипизма и полиморфизма. В отпечатках лимфоузла обнаруживаются зрелые лимфоциты и пролимфоциты;
Тотальная диффузная лимфоцитарная инфильтрация костного мозга при трепанобиопсии крыла подвздошной кости, в стернальном пунктате выраженная пролиферация зрелых лимфоцитов (около 20-40%);
Умеренный лейкоцитоз в периферической крови - около 20000-50000 в 1 мкл; в лейкоцитарной формуле около 60-80% лимфоцитов и умеренная нейтропения (количество нейтрофилов составляет до 20% и более).
Эта форма хронического лимфолейкоза носит быстропрогрессирующее течение, медиана выживаемости составляет 36 месяцев (Никитин Е.А., Лорие Ю.Ю., Меликян А.Л., 2003). Диагностика опухолевой формы ХЛЛ сразу же является основанием для назначения цитостатической терапии.
Спленомегалическая (селезеночная) форма
Спленомегалическая форма по существу была выделена уже при определении стадии хронического лимфолейкоза по Rai, когда оказалось, что стадия процесса, текущего только с лимфоцитозом и увеличением селезенки - II стадия, прогностически более благоприятна, чем все остальные, кроме нулевой, проявляющейся только лимфоцитозом в крови и костном мозге.I. Dighiero с соавт. (1979) предложили выделить селезеночную форму хронического лимфолейкоза с преимущественным увеличением селезенки при умеренном увеличении лимфатических узлов и различным уровнем лейкоцитов. Селезенка у таких пациентов может занимать большую часть брюшной полости и при прогрессировании заболевания вызывать компрессионный и болевой синдромы.
От лимфоцитомы селезенки эта форма отличается диффузным ростом лимфатических элементов в костном мозге (трепанобиоптат), лимфатических узлах, селезенке. Нередко увеличивается (не очень значительно) и печень. Количество лейкоцитов в периферической крови может быть различным, но обычно лейкоцитоз нарастает на протяжении месяцев. Часто наблюдается гемолитическая анемия.
Медиана выживаемости таких пациентов составляет 62 месяца (Никитин Е.А., Лорие Ю.Ю., Меликян А.Л., 2003).
Абдоминальная форма
Если на протяжении месяцев и лет рост опухоли ограничен почти исключительно лимфатическими узлами брюшной полости, такая форма ХЛЛ называется абдоминальной. Для выявления увеличенных внутрибрюшинных лимфоузлов используют УЗИ и КТ.Пролимфоцитарная форма
Пролимфоцитарная форма отличается, прежде всего, морфологией лимфоцитов, которые в мазках (крови и костного мозга), отпечатках имеют крупную четкую нуклеолу. Конденсация хроматина в ядре, как показывает электронная микроскопия, выражена умеренно и в основном по периферии. В гистологических препаратах лимфатических узлов и селезенки при этой форме лейкоза лимфоциты также содержат нуклеолы. Иммунологическая характеристика выявляет В- (80%), и Т-клеточную (20%) природу пролимфоцитарного лимфолейкоза.В отличие от В-лимфоцитов типичного хронического лимфолейкоза при данной форме на поверхности лейкозных лимфоцитов обнаруживается обилие иммуноглобулинов, чаще М - или D-типа; кроме того, эти лимфоциты образуют мало розеток с эритроцитами мыши. По выражению А.И Воробьева и соавт. (2003), «по характеристике иммунологических маркеров пролимфоцитарная форма старше обычного злокачественного лимфолейкоза». При этой форме ХЛЛ очень часто определяют хромосомные аномалии.
Основными клинико-лабораторными особенностями пролимфоцитарной формы являются:
Возраст больных в 50% случаев старше 70 лет;
выраженная слабость, похудание, наклонность к геморррагиям;
значительная спленомегалия
не выраженное увеличение лимфатических узлов;
инфильтрация лейкемическими клетками кожи в области туловища, лица, рук, появление папулезной, незудящейся сыпи (приблизительно у 1/3 больных с Т-пролимфоцитарным лейкозом;
Изменения в анализе периферической крови: у 70-80% больных количество лимфоцитов превышает 100000 в 1 мкл (100х10 9 /л), при этом 30-50% всех лимфоцитов представлены пролимфоцитами; характерны анемия и тромбоцитопения;
положительная цитохимическая реакция лимфоидных клеток на активность кислой фосфотазы (в форме гранул, иногда формирующих блок, полностью подавляется тартратом); около половины клеток содержит а-нафтилэстеразу; в лимфоидных клетках обнаруживается гликоген в виде мелких гранул; реакция на миелопероксидазу отрицательная;
выраженная пролиферация пролимфоцитов в костном мозге (по данным миелограммы);
быстрое прогрессирование заболевания и низкая эффективность цитостатической терапии.
Средняя продолжительность жизни пациентов с пролимфоцитарной формой ХЛЛ около 3 лет.
Костномозговая форма
Впервые была описана в 1937 г. Storti под названием limfadenia ossium. Для этой формы характерны быстро прогрессирующая панцитопения, тотальное или частичное замещение костного мозга диффузно растущими зрелыми лимфоцитами. Лимфатические узлы не увеличены, селезенка за очень редким исключением также не увеличена, печень нормальных размеров.Морфологически отмечается гомогенность структуры ядерного хроматина, иногда его пикнотичность, реже имеются элементы структурности, отдаленно напоминающие бластную; цитоплазма с выраженной базофилией, узкая, часто обрывчатая. Прогноз - крайне неблагоприятный.
В качестве примера приводим два случая, типичного классического течения хронического лимфолейкоза и редкой формы этого заболевания.
Клинические примеры
Больной А., 1933 г.р. Диагноз «Хронический лимфолейкоз» был впервые выставлен в 1991г. При первичной диагностике имела место стадия А по классификации Binet. Периферические лимфоузлы не более 2см в диаметре. На коже и видимых слизистых геморрагического синдрома нет. В легких везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. Живот мягкий, безболезненный при пальпации. Печень по краю реберной дуги. Селезенка не увеличена.Клинический анализ крови: гемоглобин - 140 г/л, эритроциты - 4,5 х 10 12 /л, лейкоциты - 27х10 9 /л, тромбоциты - 180х10 9 /л, сегментоядерные - 12%, моноциты - 2%, лимфоциты 85%.
С 1991 по 1996 гг. за больным велось динамическое наблюдение в условиях поликлиники. В 1996 г. отмечено увеличение лимфатических узлов до 2 см в диаметре. Нижний полюс селезенки стал пальпироваться в левом подреберии. Была назначена первично-сдерживающая терапия хлорамбуцилом. С 1996 по 2005 г. на фоне первично-сдерживающей терапии прогрессии заболевания не отмечалось.
С 2005 г. в связи со значительным увеличением селезенки (нижний полюс пальпировался на уровне тазовых костей) и печени (нижний край пальпировался на 12 см ниже края реберной дуги), генерализованной лимфаденопатией (лимфатические узлы увеличены до 4-5 см, плотноэластической консистенции, спаяны в конгломераты), стали проводить курсовую цитостатическую терапию (монотерапия циклофосфаном, курсы полихимиотерапии по протоколам СР, СОР, СНОР).
С 2008 года начата терапия по протьоколу FCR, проведено 8 курсов (2008-2009 гг). Перед началом терапии по протоколу FCR размеры периферических лимфатических узлов всех групп до 3-4 см, спаяны в конгломераты. По данным КТ, увеличены медиастинальные, внутрибрюшин-ные и забрюшинные лимфоузлы, спаяны в конгломераты. Печень - нижний край пальпировался на 10 см ниже края ребернгой дуги.
Нижний полюс селезенки пальпировался на уровне пупка. Клинический анализ крови: гемоглобин - 65 г/л, эритроциты - 2,5х10 9 /л, лейкоциты - 120х10 9 /л, тромбоциты - 80х10 9 /л, сегментоядерные - 2%, лимфоциты 98%. После проведения 8 курсов RFC была достигнута полная ремиссия ХЛЛ. Лимфоузлы всех групп в пределах нормы. Геморрагического синдрома на коже и видимых слизистых нет. В легких везикулярное дыхание, хрипов нет. Живот мягикий, безболезненный при пальпации. Печень по краю реберной дуги. Селезенка пальпаторно не определялась.
Ремиссия сохранялась с 2009 по 2014 гг. В ноябре 2014 г. - рецидив. Периферические лимфоузлы всех групп увеличены до 3-4 см, спаяны в конгломераты, каменистой плотности. Нижний полюс селезенки пальпируется на уровне тазовых костей. По данным КТ увеличены медиастинальные, внутрибрюшинные и забрюшинные лимфоузлы.
Клинический анализ крови: гемоглобин - 95 г/л, эритроциты - 3,0х10 12 /л, лейкоциты - 17х10 9 /л, тромбоциты - 100х10 9 /л, сегментоядерные - 5%, лимфоциты 95%. По данным гистологического исследования лимфоузла диагностирована трансформация в крупноклеточную лимфому (синдром Рихтера).
Больной К., 32 года, впервые поступил в гематологическое отделение Амурской областной клинической больницы в ноябре 2010г. При прохождение мед. осмотра в гемограмме выявлены изменения - лейкоцитоз, лимфоцитоз, клинически - спленомегалия. Больной направлен на консультацию к гематологу в Амурскую областную консультативную поликлинику. Пациент госпитализирован в гематологическое отделение с целью обследования и определения дальнейшей тактики ведения.
При поступлении: состояние стабильное, жалобы на незначительную слабость. Кожный покров чистый, обычной окраски, геморрагического синдрома нет. Периферические лимфоузлы не увеличены В легких дыхание везикулярное, ослабленное в верхних отделах слева, единичные сухие хрипы, частота ударов (ЧД) 18 в мин. Тоны сердца ритмичны, приглушены, частота сердечных сокращений (ЧСС) 78 в мин, артериальное давление (АД) 120/80 мм.рт.ст. Живот мягкий безболезненный, печень по краю реберной дуги, селезенка - занимала большую часть живота: нижний полюс пальпировался на уровне тазовых костей, правая граница - от пупка вправо на 10 см. Стул и диурез в норме.
В клиническом анализе крови при первичном поступлении от 23.11.10г.: гемоглобин - 112г/л; эритроциты - 3,54х10 12 /л; лейкоциты - 156,6х10 10 /л; тромбоциты - 90х10 9 /л; скорость оседания эритроцитов (СОЭ) - 50 мм/ч, пролимоциты - 77%, сегментоядерные- 3%, лимфоциты - 20%.
Б/х анализ крови от 23.11.10: глюкоза - 6,1 ммоль/л; билирубин - 16,6-13,2-3,4 мкмоль/л; мочевина - 4,7 ммоль/л; белок - 70г/л, АСТ - 64МЕ/л, аланинаминотрансфераза (АЛТ) - 34МЕ/л, тимоловая проба-2,0, фибриноген-3,0 г/л, протромбиновый индекс (ПТИ) - 55%.
Иммуноферментный анализ (ИФА) на гепатиты от 23.11.10: отрицательный. Кровь на RW от 23.11.10: отрицательный. Кровь на ВИЧ от 23.11.10: отрицательный.
Миелограмма от 23.11.10 №80: получен гиперклеточный костный мозг, регенерация мегакариоцитарного ростка, увеличение лимфоидных элементов - 34,6% (18,6% пролимфоциты)
Иммунофенотипирование от 25.11.10: CD3-15,3%; CD3CD4-49,6%; CD5-27,0%; CD8-41,1%; CD4/CD8-1,2%; CD10-33,8%; CD19-62,0%; CD20-69,7%; CD22-43,7%; CD23-4,0%; CD25-48,3%; CD38-18,9%; HLA-DR-60,9.
УЗИ внутренних органов от 27.11.10: Гепатоспленомегалия (печень - ПД 160мм, ЛД 95 мм, портальная вена - 12 мм; селезенка - около 300х128 мм, селезеночная вена - 11 мм), селезенка гигантских размеров. Уплотнение по ходу портальных трактов печени. Диффузные изменения паренхимы поджелудочной железы.
Трепанобиопсия подвздошной кости от 25.11.10 №21421: среди костных балок участки костного мозга, в которых соотношение кроветворного костного мозга и жировой ткани 95:5. Диффузная инфильтрация зрелыми лимфоцитами. Мегакариоциты единичные.
Учитывая данные проведенного исследования, пациенту был выставлен диагноз: Лимфома селезёнки с лейкемизацией.
6.12.2010г выполнена спленэктомия. Перенёс удовлетворительно. Гистологические препараты (блоки) были направлены в РОНЦ им.Блохина. Гистология 1639/11 (РОНЦ им. Блохина): в гистологических препаратах, изготовленных из предоставленных блоков, в срезах ткани селезенки гистоархитектоника нарушена за счет массивного диффузного инфильтрата из мономорфных небольших лимфоидных клеток с округлыми ядрами, в которых визуализируется маленькое ядрышко.
В сосудах капиллярного и синусоидного типа - мелкие лимфоидные клетки с вышеописанной морфологией. На срезах с парафинового блока проведено иммуногистохимию (ИГХ) - исследование с испотльзованием антител. Клетки диффузного опухолевого инфильтрата мономорфно экспрессируют СД20, Jg M. С другими маркерами в опухолевых клетках реакции негативны. Среди опухолевого субстрата видны скопления Т-клеток СД2+, СД3+, СД4+, СД5+, СД8+ (лимфоидные, литториальные клетки). Заключение: С учетом клинико-лабораторных данных, морфологических особенностей и иммунофенотипа в ткани селезенки поражение субстратом В-клеточного пролимфоцитарного лейкоза.
По полученным результатам иммуногистохимического исследования пациенту выставлен диагноз: В-клеточный пролимфоцитарный лейкоз. Пациенту проведено 6 курсов полихимиотерапии (ПХТ) по протоколу «FCR». Последний курс в августе 2011г. Достигнута полная ремиссия заболевания, которая сохраняется по настоящее время.
В настоящее время (апрель 2015 г) состояние удовлетворительное. Периферические лимфоузлы всех групп не более 1 см. в диаметре. По данным КТ лимфоузлы средостения, забрюшинные и внутрибрюшинные не увеличены. В легких везикулярное дыхание, хрипов нет. Тоны сердца ясные, ритм правильный. Живот мягкий, безболезненный при пальпации. Печень не увеличена. Клинический анализ крови от апреля 2015г.: гемоглобин - 151 г/л; эритроциты - 5,02х10 12 ; лейкоциты - 8,5х10 12 ; тромбоциты - 365х 10 9 ; СОЭ - 4мм/ч, сегментоядерные - 43%, эозинофилы - 8%, моноциты - 8%, лимфоциты - 41%.
Терминальная стадия
Терминальная стадия ХЛЛ характеризуется резким прогрессирующим ухудшением общего состояния больных, истощением, выраженной интоксикацией, исчезновением аппетита, высокой температурой тела. Гипертермия может быть обусловлена не только самим ХЛЛ, нередко она связана с развитием туберкулеза легких или присоединением тяжелой бактериальной пневмонии.Для терминальной стадии характерно развитие тяжелых осложнений. В первую очередь это инфекционно-воспалительные процессы с локализацией в различных органах и системах. Тяжелая генерализованная инфекция часто является причиной смерти больных ХЛЛ.
Течение хронического лимфолейкоза могут осложнять пневмококковая, стрептококковая пневмония, герпетическая инфекция и др. Возможны инфекционные поражения не только легких, но и мочевыводящих путей, кожи. Развитию инфекционно-воспалительных процессов при ХЛЛ способствуют нарушения иммунной системы, гипогаммаглобулинемия.
Герпетическая инфекция может осложнить течение ХЛЛ на любой стадии, но особенно в терминальной фазе заболевания. Чаще это опоясывающий лишай, у многих больных вирус герпеса может вызвать генерализованное поражение кожи, слизистых оболочек полости рта, ЖКТ, мочеполовой системы.
Одним из грозных клинических признаков терминальной фазы ХЛЛ является тяжелая почечная недостаточность. Она обусловлена инфильтрацией почечной ткани лейкозными клетками и проявляется развитием олигоанурии со значительным повышением в крови содержания мочевины, креатинина, остаточного азота. Тяжелая почечная недостаточность может послужить причиной смерти больных ХЛЛ.
В терминальной фазе хронического лимфолейкоза возможно развитие нейролейкемии в связи с интенсивной инфильтрацией мозговых оболочек молодыми лимфоцитами. Клиническая картина нейролейкемии при ХЛЛ соответствует таковой при остром лейкозе и проявляется сильными головными болями, рвотой, развитием менингеального синдрома, парезов черепномозговых нервов, периферическими параличами. Лейкозная инфильтрация корешков спинномозговых нервов сопровождается интенсивными «корешковыми» болями.
В терминальной фазе в связи с лимфоидной инфильтрацией могут развиваться тяжелая кардиопатия, приводящая к недостаточности кровообращения, поражение легких с симптоматикой выраженной дыхательной недостаточности, экссудативный плеврит (его необходимо дифференцировать с экссудативными плевритом туберкулезного генеза).
Характерным признаком терминальной стадии ХЛЛ является выраженная анемия. Она обусловлена сокращением красного кроветворного ростка в связи с лимфоидной инфильтрацией костного мозга, интоксикацией и аутоиммунными механизмами. Характерны также тромбоцитопения и усиление явлений геморрагического диатеза.
У некоторых больных в терминальной стадии развивается бластный криз, но чаще наблюдается трансформация в другие лимфопролиферативные заболевания. В терминальном периоде ХЛЛ отмечается значительное прогрессирующее увеличение лимфатических узлов и селезенки. Обычно это свидетельствует о трансформации ХЛЛ в злокачественные лимфопролиферативные заболевания. Известно, что хронический лимфолейкоз может трансформироваться в синдром Рихтера, в пролимфоцитарный лейкоз, острый лимфобластный лейкоз, плазмаклеточный лейкоз, миеломную болезнь.
Синдром Рихтера
Синдром (трансформация) Рихтера - это переход ХЛЛ в диффузную, агрессивную крупноклеточную иммунобластную лимфому, состоящую из крупных В-лимфоцитов. Описан в 1928 г. Наблюдается у 3-10% больных с прогрессирующей формой ХЛЛ. В развитии этого синдрома большая роль принадлежит мутации гена р53.Основными клиническими признаками синдрома Рихтера являются (Robertson и соавт., 1993; Османов Д.Ш., 2007):
Прогрессирующее увеличение лимфатических узлов, они приобретают каменистую плотность, инфильтрируют и сдавливают соседние ткани, в том числе и лимфатические пути, что вызывает появление болей и отека; у многих больных развивается ретроперитонеальная лимфаденопатия;
Повышение температуры тела;
Уменьшение массы тела;
Увеличение степени гепатомегалии;
Развитие массивной спленомегалии;
Появление неврологической симптоматики вследствие вовлечения в патологический процесс ЦНС;
Клинические признаки выраженной инфильтрации опухолевыми клетками других органов - желудочно-кишечного тракта (ЖКТ) , легких, почек;
Высокий уровень в сыворотке крови лактатдегидрогеназы и моноклоновая гаммапатия (выявляется при электрофорезе белков).
Диагноз рихтеровской трансформации уточняется биопсией лимфатических узлов. Характерным является наличие в биоптатах больших иммунобластных клеток, которые имеют выраженную базофильную цитоплазму и ядро неправильной формы с нуклеолами. В костном мозге обнаруживается инфильтрация этими незрелыми клетками, которые могут вызывать очаги остеолиза.
Продолжительность жизни больных с синдромом Рихтера составляет около 6-12 месяцев (Османов Д.Ш., 2007).
Трансформация в пролимфоцитарный лейкоз
Приблизительно у 15% больных В-клеточным хроническим лимфолейкозом в процессе опухолевой прогрессии кроме малых лимфоцитов в периферической крови могут появляться пролимфоциты, количество которых может достигать от 10 до 90 %. Это свидетельствует о трансформации ХЛЛ в пролимфоцитарный лейкоз.Клиническая картина при этом в целом соответствует клинической картине ХЛЛ, но отличительным признаком является прогрессирующая, очень выраженная спленомегалия. У некоторых больных обнаруживается транслокация t(6;12). Больные хроническим лимфолейкозом с трансформацией в пролимфоцитарный лейкоз значительно хуже реагируют на проводимую химиотерапию, у них сокращается продолжительность жизни. По данным Chani и соавт. (1986), после трансформации продолжительность жизни больных ХЛЛ составляет 9 месяцев.
Трансформация в острый лейкоз
Очень редко, приблизительно в 2% случаев, наступает трансформация ХЛЛ в острый лимфобластный лейкоз. Установлено, что при этом лейкозный клон происходит из того же клона В-клеточной направленности, что и при ХЛЛ. Бластная трансформация ассоциируется с увеличением экспрессии гена с-MYC и генов, отвечающих за синтез иммуноглобулинов.Лейкемические бластные клетки экспрессируют терминальную дезоксинуклеотидтрансферазу (TdT) , высокий уровень поверхностного иммуноглобулина и главного комплекса гистосовместимости (HLA-DR) . Клинические проявления заболевания соответствуют клинике острого лимфобластного лейкоза.
В редких случаях наблюдается трансформация ХЛЛ во множественную миелому. При этом предполагается существование отдельного патологического клона плазматических клеток в костном мозге.
В.В. Войцеховский, Т.В. Заболотских, С.С. Целуйко, Ю.С. Ландышев, А.А. Григоренко
Вам также будет интересно:

Выбор системы налогообложения осуществляется по заявлению индивидуального предпринимателя....

ИП, пожелавшие вести деятельность при уплате единого специального налога на вмененный...

Предприниматель Александр Харченко поделился с сайт опытом открытия ИП через интернет. Если...

Города Древней Руси
Зарождение городов в Древней Руси относится к IX-X вв. Их возникновение...
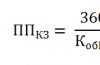
К основным видам кредиторской задолженности относятся: перечисление денежных средств на...

